
Performance evaluations
Performance evaluations
-
GROMACS
Three benchmark datasets are used to evaluate performance: water molecules (synthetic), Lignocellulose (polymer), and hEGFR protein dimers/tetramers from HECBioSim. Also, two compiler sets, GCC 13.1.0/openMPI 4.2.5 and Intel oneAPI 2023.0.0 are used to compile GROMACS. In Figure 4, Figure 5, and Figure 6 compilers are color-coded while the datasets are represented by marker shapes. Intel oneAPI performance is plotted in orange while GCC/openMPI performance is represented blue.
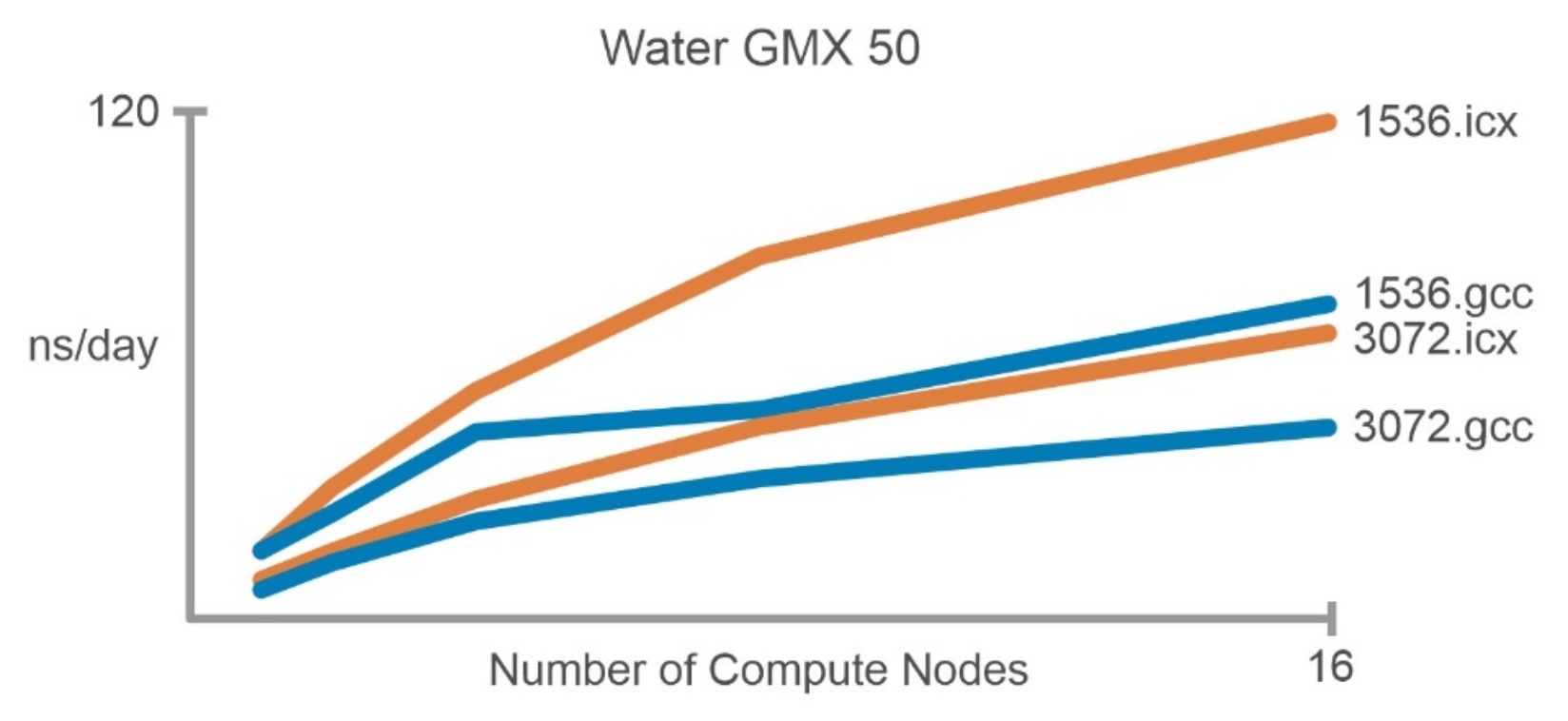 Figure 4. Simulation performance comparisons with Water GMX50 and two compilers
Figure 4. Simulation performance comparisons with Water GMX50 and two compilers For water data, optimized Intel oneAPI compiled GROMACS performs nearly two times better than the GCC/openMPI version for both 1.5 million and 3 million water molecules as shown in Figure 4.
Lignocellulose is a plant biopolymer that consists of cellulose, hemicellulose, and lignin. This is where GCC/openMPI compiler performs better than Intel, as shown in Figure 5. Figure 5. Simulation performance comparisons with Lignocellulose and two compilers
Figure 5. Simulation performance comparisons with Lignocellulose and two compilers
Other real-life datasets are obtained from HECBioSim. This hEGFR protein forms polymers naturally. Therefore, dimer (1.4 million atoms) and tetramer (3 million atoms) datasets are tested here. The plot in Figure 6 shows a similar performance boost with the Intel oneAPI compiler. Figure 6. Simulation performance comparisons with hEGFR protein multimers and two compilers
Figure 6. Simulation performance comparisons with hEGFR protein multimers and two compilersThe complexity of biopolymers is not constant and seems to affect the simulation performance. We recommend performing compiler-based optimizations for various data since the performance differences should not be ignored, as shown in this study.
BWA-GATK v3.6 germline variant calling pipeline
This pipeline performs germline variant calling, and the performance of the pipeline is measured in runtime (hours) and genomes per day.
Single sample performance
A single 50x WGS (ERR194161) is processed with 8480+, one of the 4th Generation Intel® Xeon® Scalable Processors in the PowerEdge C6620 server. All the cores in this two-socket system were used to run BWA-GATK v3.6 pipeline although it is not an optimal way to run this pipeline. As shown in Figure 7, the base frequency of the CPU is a crucial factor for the speedy process. However, using more than 12 cores for the applications in this pipeline becomes extremely inefficient because the applications are not scaling well. Generally, it is better to maximize throughput rather than minimizing turnaround time unless a short turnaround time is mission-critical.
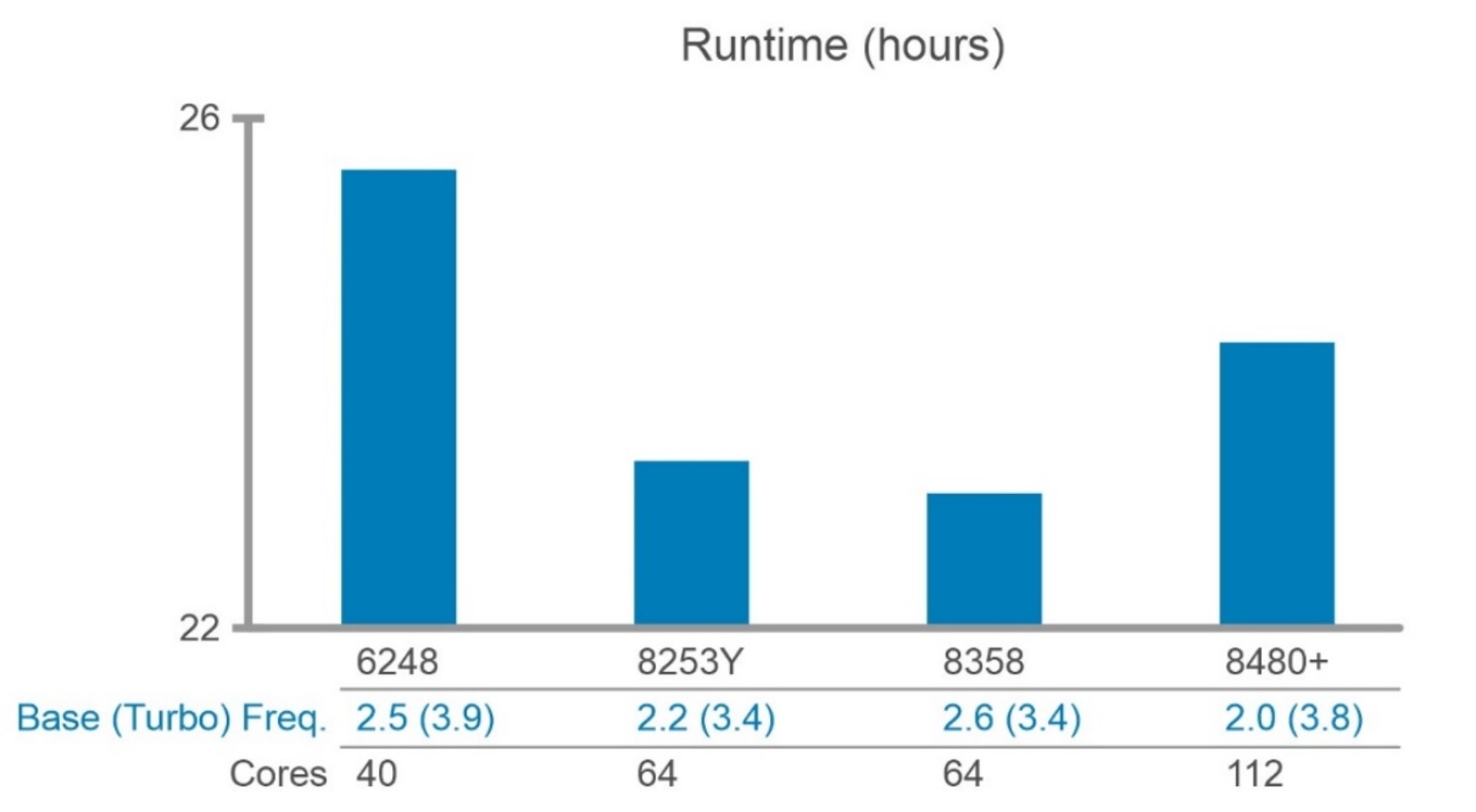 Figure 7. BWA-GATK v3.6 performance with a single 50x WGS and a single node
Figure 7. BWA-GATK v3.6 performance with a single 50x WGS and a single nodeThroughput of a single node: multi-sample/single node performance
As the number of samples processed concurrently is increased (x-axis in Figure 8), the runtime to complete multiple pipelines increases (left y-axis in Figure 8). With 32 samples processed on a single compute node in 68.13 hours (about 3 days), genomes per day are calculated to be 11.27 (right y-axis in Figure 8). This is the maximum capacity of a single C6620 with an Intel 8480+ CPU.
 Figure 8. BWA-GATK v3.6 performance with multi-sample and single C6630 server
Figure 8. BWA-GATK v3.6 performance with multi-sample and single C6630 serverThroughput of multiple nodes: multi-sample and multi-node performance
From the multi-sample and single-node performance study, 32 samples per C6620 is the maximum number of samples that can be processed together. However, the number of samples per C6620 is set to 20 due to the storage capacity limitation with the current PowerScale F600 configured for this study.
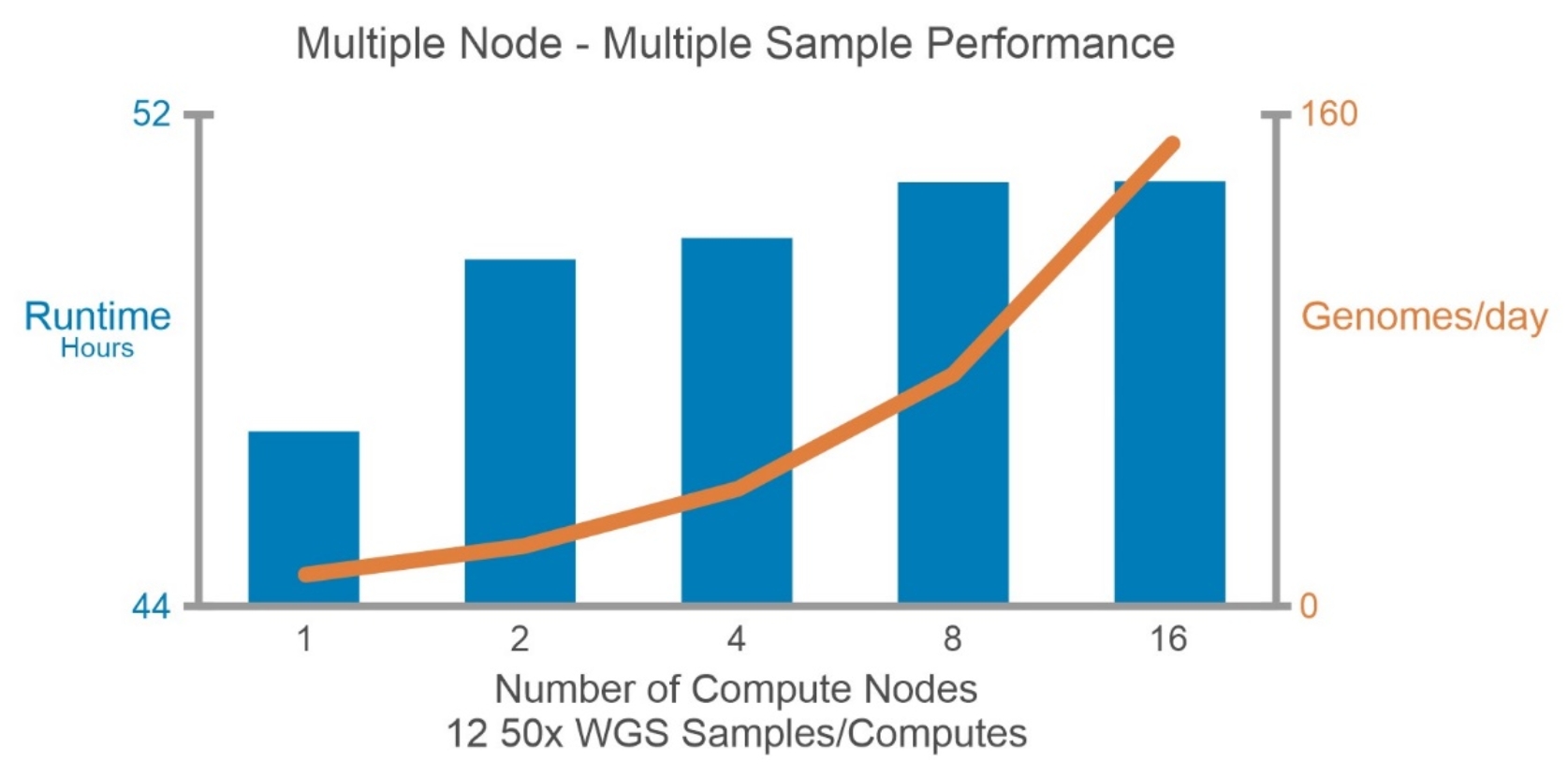 Figure 9. BWA-GATK v3.6 performance with multi-sample and multi-node performance
Figure 9. BWA-GATK v3.6 performance with multi-sample and multi-node performance