
Introduction
Introduction
-
Life science, the study of life, is an extremely broad term that encompasses many different disciplines. Life science is a more recent development that has been fueled by the growth of technology and the advancement of scientific knowledge, while biology has been around for centuries. Our validated systems evolved from the Genomics Data Analysis Platform (GDAP) about nine years ago. GDAP was an optimized system to perform a variant calling pipeline for human WGS data. It was relatively simple to optimize for a single pipeline with few applications compared to the current DVD for HPC Life Sciences which is validated with an extended benchmark portfolio with differentially expressed gene (DEG) study for RNA-seq data, De Novo Assembly, MDS, and Cryo-EM.
Traditionally, life sciences hardly fit into the HPC realm in terms of data size. Like other scientific areas, simulation such as MD simulation, neural simulation, epidemic simulation, and others, was the major application. But with technological breakthroughs in sequencing, microscopy, and imaging, there has been explosive data growth in genomics, cryo-electron microscopy (cryo-EM), and digital imaging. Both NGS and cryo-EM are becoming mainstream technologies for studying genes, the architecture of cells, and protein assemblies.
Next Generation Sequencing (NGS)
NGS changed the paradigm in the genomics field by allowing us to observe the entire genes simultaneously and at a lower cost. As NGS sequencing platforms continue to drive down the costs for a whole human genome, it is now playing an increasingly important role in clinical practice for patient care along with being a critical tool for public health initiatives. The major market leader Illumina, which controls about 80 percent of the global DNA sequencing market, unveiled the NovaSeq X series. This new series will reduce the cost to $200 per human genome while providing results at twice the speed of the previous generation. One of the high-throughput models, the NovaSeq X Plus system could generate around 16 TB of sequencing data every other day. Furthermore, Complete Genomics Inc. and MGI Tech Co. announced under $100 for WGS.
Types of NGS data
All types of NGS share a similar sequencing workflow, but there are many types of NGS. As shown in Figure 1, typical NGS applications are summarized based on the sequencing targets and purposes. These applications fall into two major groups, DNA-Seq and RNA-Seq. Most of the DNA-Seq studies are focused on genomic mutations or Single Nucleotide Polymorphism (SNP) in chromosomes while RNA-Seq studies provide snapshots of the entire gene expressions at a given point in time.
The information encoded in an individual's genome is the foundation of precision medicine, driving diagnosis and supporting therapeutic decisions for disease treatment and in some cases prevention strategies due to person-to-person variability. For a typical DNA-Seq, identifying genetic variants or differences within a genome is done by comparing an individual’s genome to a genome reference. Alternatively, RNA-Seq rapidly replaces microarray technology and becomes a standard procedure for differentially expressed gene and RNA-Profiling studies. The ability to perform DNA-Seq and RNA-Seq at the same time for the same sample opened a new opportunity in the translational research area. Identifying mutations from genes in chromosomes and knowing how those mutations change the gene expressions helps to complete the puzzle from the central dogma. Although we still do not have comprehensive protein information, it is much easier to investigate a few target proteins instead of testing many proteins. Currently, there are various projects to label genomic variations in humans according to their disease type, phenotypic characteristics, drug treatments, clinical outcomes, and so on. These data integration efforts will eventually provide the basis for precision medicine and personalized medicine.
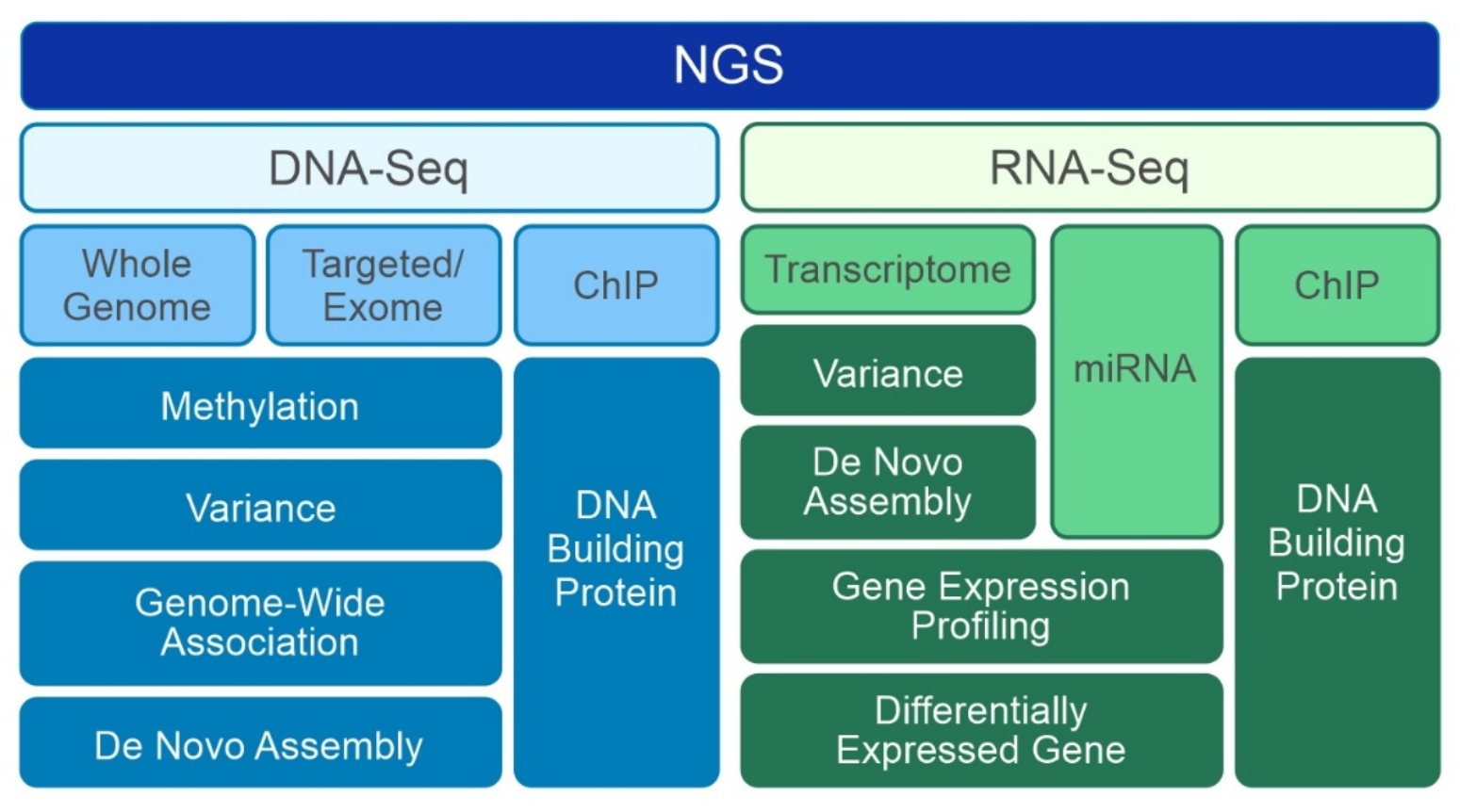 Figure 1. Types of NGS
Figure 1. Types of NGSDepth of coverage
When planning time and resources to complete NGS secondary analysis, it is essential to understand the depth of coverage for sample data as it will impact analysis time per sample. The depth of coverage describes the average number of reads in sequencing that align to, or cover, a known reference sequence. Although equal sequencing coverage does not guarantee equal scientific value, higher coverage is important to ensure the alignment observations are statistically sound.
The depth of coverage requirements varies by sequencing application. For example, 30x to 60x coverage is common for human WGS data. However, the analysis of cancer genomes may require sequencing to a depth of coverage higher than 100x to achieve the necessary sensitivity and specificity to detect rare, low-frequency variants.
The depth of coverage is also a measure of the amount of data per sample. As coverage increases, so does the amount of data per sample. For example, a 50x (coverage) WGS sample contains approximately five times more data than a 10x WGS sample, meaning secondary analysis time will also increase proportionally to the amount of data.
Working with NGS data
There are three main steps in the NGS data analysis process. The first step, primary analysis, is tightly coupled with the sequencing step. During cycles of chemical reactions and imaging, real-time analysis software provides base calls that can be constructed for millions of short DNA or RNA fragments. Primary analysis is typically built into a sequencer. Once millions of DNA or RNA fragments are generated from primary analysis, these fragments need to be aligned or assembled to be useful since the positional information was lost during the sequencing procedure. The fragments with the original position information can be used to study various aspects listed in Figure 1. For this reason, these steps are called secondary analysis. After secondary analysis, the next step is tertiary data analysis which includes biological data mining, statistical analysis, biological interpretation, and so on. Tertiary analysis can convert data into knowledge.
The characteristics of NGS data in terms of their workflows are unique. Secondary analysis is mostly parallel and includes read cleanup, alignment, variant calling, and gene expression analysis. Although each sample in a study can be processed independently, it must be processed the same way for tertiary analysis. The results from the various secondary analyses cannot be mixed for a subsequent tertiary analysis due to the strong association in a sample set or cohort in these biological samples. If NGS data for different studies or cohorts is regrouped, the regrouped samples must be reprocessed with a specific secondary analysis. Due to this, the sequencing data often goes through multiple secondary analysis processes.
Reducing analysis time
A time-consuming aspect of secondary analyses is WGS. WGS generates a list of genetic variants and can take minutes to days depending on the size of the dataset coupled with the available software, computing, and storage resources. Due to the size of individual sample data and volume of samples, WGS secondary analysis is a compute and storage-intensive process. The most used and cited methods for secondary analysis include the Burrows-Wheeler Alignment (BWA-Mem) (Li, 2009), and the Genome Analysis Tool Kit (GATK) (McKenna, 2010). Using the Broad GATK Best Practices workflow (pipeline) requires over 30 hours to process 40x WGS (Goyal, 2017). The most recent test results in Dell Technologies as part of the endeavor to build a DVD show roughly 24 hours to process 50x WGS with dual Intel Xeon Platinum 8480+ Processors. Analyzing a few genomes per day is far from ideal when a modern, high throughput NGS instrument can generate unanalyzed, raw NGS data for 20 or more WGS within 24 hours.
It is important to consider all the critical variables that may impact the total secondary analysis (wall clock) time when choosing technologies that enable secondary analysis of NGS data. These variables are wide-ranging and entail the type of NGS sequencing application including the sequencing coverage per sample, supporting analysis software and strategies that are specific to the application, output file types, application file access patterns, and number and type of available computing resources.