




HPC Application Performance on Dell PowerEdge R6625 with AMD EPYC- GENOA
The AMD EPYC 9354 Processor, when integrated into the Dell R6625 server, offers a formidable solution for high-performance computing (HPC) applications. Genoa, which is built on the Zen 4 architecture, delivers exceptional processing power and efficiency, making it a compelling choice for demanding HPC workloads. When paired with the PowerEdge R6625's robust infrastructure and scalability features, this CPU enhances server performance, enabling efficient and reliable execution of HPC applications. These features make it an ideal choice for HPC application studies and research.
At Dell Technologies, it’s our goal to help accelerate time to value for our customers. Dell wants to help customers leverage our benchmark performance and scaling studies to help plan out their environments. By utilizing our expertise, customers don’t have to spend time testing different combinations of CPU, memory and interconnect or choosing the CPU with the sweet spot for performance. This also saves time, as customers don’t have to spend time deciding which BIOS features to tweak for best performance and scaling. Dell wants to accelerate the set-up, deployment, and tuning of HPC clusters to enable customers to get the real value- running their applications and solving complex problems like manufacturing better products for their customers.
Testbed configuration
Benchmarking for high-performance computing applications was carried out using Dell PowerEdge 16G servers equipped with AMD EPYC 9354 32-Core Processor.
Table 1. Test bed system configuration used for this benchmark study
Platform | Dell PowerEdge R6625 |
Processor | AMD EPYC 9354 32-Core Processor |
Cores/Socket | 32 (64 total) |
Base Frequency | 3.25 GHz |
Max Turbo Frequency | 3.75 GHz |
TDP | 280 W |
L3 Cache | 256 MB |
Memory | 768 GB | DDR5 4800 MT/s |
Interconnect | NVIDIA Mellanox ConnectX-7 NDR 200 |
Operating System | RHEL 8.6 |
Linux Kernel | 4.18.0-372.32.1 |
BIOS | 1.0.1 |
OFED | 5.6.2.0.9 |
System Profile | Performance Optimized |
Compiler | AOCC 4.0.0 |
MPI | OpenMPI 4.1.4 |
Turbo Boost | ON |
Interconnect | Mellanox NDR 200 |
Application | Vertical Domain | Benchmark Datasets |
OpenFOAM | Manufacturing - Computational Fluid Dynamics (CFD) | Motorbike 50M 34M and 20M cell mesh |
Weather Research and Forecasting (WRF) | Weather and Environment |
|
Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) | Molecular Dynamics | Rhodo, EAM, Stilliger Weber, tersoff, HECBIOSIM, and Airebo |
GROMACS | Life Sciences – Molecular Dynamics | HECBioSim Benchmarks – 3M Atoms , Water and Prace LignoCellulose |
CP2K | Life Sciences | H2O-DFT-LS-NREP- 4,6 H2O-64-RI-MP 2 |
Performance Scalability for HPC Application Domain
Vertical – Manufacturing | Application - OPENFOAM
OpenFOAM is an open-source computational fluid dynamics (CFD) software package renowned for its versatility in simulating fluid flows, turbulence, and heat transfer. It offers a robust framework for engineers and scientists to model complex fluid dynamics problems and conduct simulations with customizable features. In this study, worked on OpenFOAM version 9, which have been compiled with gcc 11.2.0 with OPENMPI 4.1.5. For successful compilation and optimization on the AMD EPYC processors, additional flags such as ' -O3 -znver4' have been added.
The tutorial case under the simpleFoam solver category, motorBike, has been used to evaluate the performance of OpenFOAM package on AMD EPYC 9354 processors. Three different types of grids were generated such as 20M, 34M, and 50M cells using the blockMesh and snappyHexMesh utilities of OpenFOAM. Each run was conducted with full cores (64 cores per node) and from single node to sixteen nodes, The scalability tests were done for all the three sets of grids. The steady state simpleFoam solver execution time was noted down as performance numbers. The figure below shows the application performance for all the datasets.
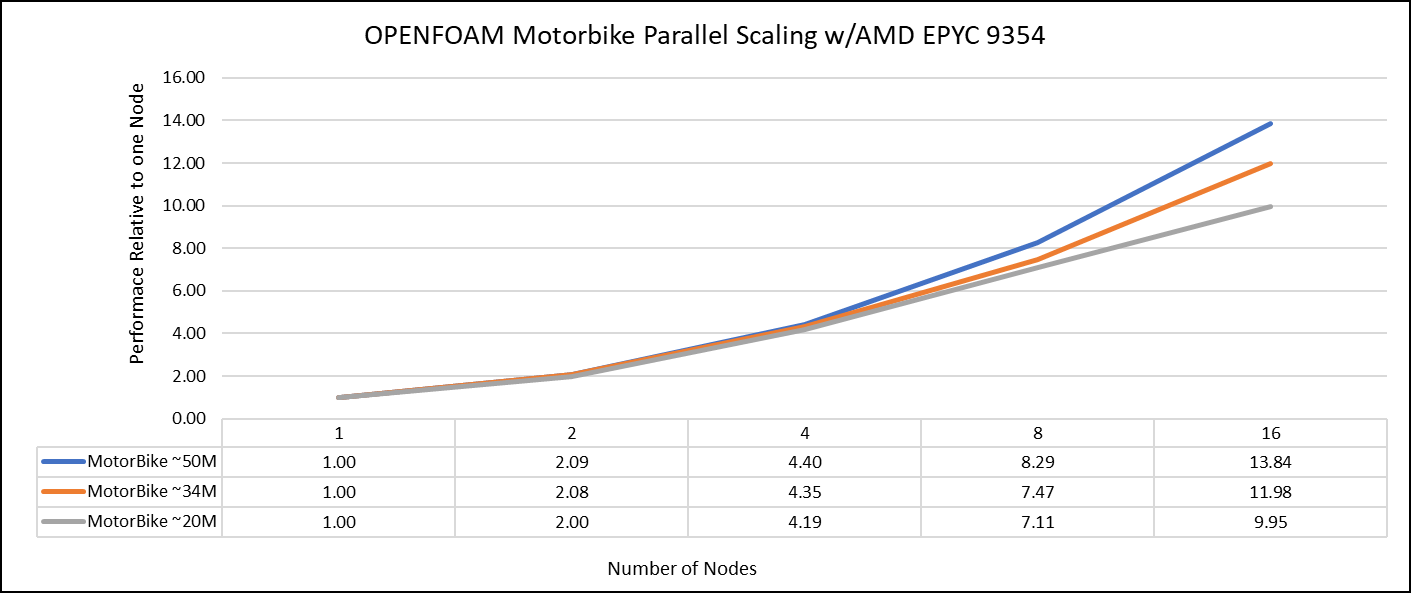
Figure 1: The scaling performance of the OpenFOAM Motorbike dataset using the AMD EPYC Processor, with a focus on performance compared to a single node.
The results are non-dimensionalized with single node result. The scalability is depicted in Figure 1. The OpenFOAM application shows linear scaling from a single node to eight nodes on 9354 processors for higher dataset (50M). For other smaller datasets with 20M and 34M cells, the linear scaling was shown up to four nodes and slightly scaling reduced on eight nodes. For all the datasets (20M, 34M and 50M) on sixteen nodes the scalability was reduced.
Achieving satisfactory results with smaller datasets can be accomplished using fewer processors and nodes, because smaller datasets do not require a higher number of processors. Nonetheless, augmenting the node count, and therefore, the processor count, in relation to the solver's computation time leads to increased interprocessor communication, subsequently extending the overall runtime. Consequently, higher node counts are more beneficial when handling larger datasets within OpenFOAM simulations.
Vertical – Weather and Environment | Application - WRF
The Weather Research and Forecasting model (WRF) is at the forefront of meteorological and atmospheric research, with its latest version being a testament to advancements in high-performance computing (HPC). WRF enables scientists and meteorologists to simulate and forecast complex weather patterns with unparalleled precision. In this study, we have worked on WRF version 4.5, which have been compiled with AOCC 4.0.0 with OPENMPI 4.1.4. For successful compilation and optimization with the AMD EPYC compilers, additional flags such as ' -O3 -znver4' have been added.
The dataset used in our study is CONUS v4.4. This means that the model's grid, parameters, and input data are set up to focus on weather conditions within the continental United States. This configuration is particularly useful for researchers, meteorologists, and government agencies who need high-resolution weather forecasts and simulations tailored to this specific geographic area. The configuration details, such as grid resolution, atmospheric physics schemes, and input data sources, can vary depending on the specific version of WRF and the goals of the modeling project. In this study, we have predominantly adhered to the default input configuration, making minimal alterations or adjustments to the source code or input file. Each run was conducted with full cores (64 cores per node) and from single node to sixteen nodes. The scalability tests were conducted and the performance metric in “sec” was noted.
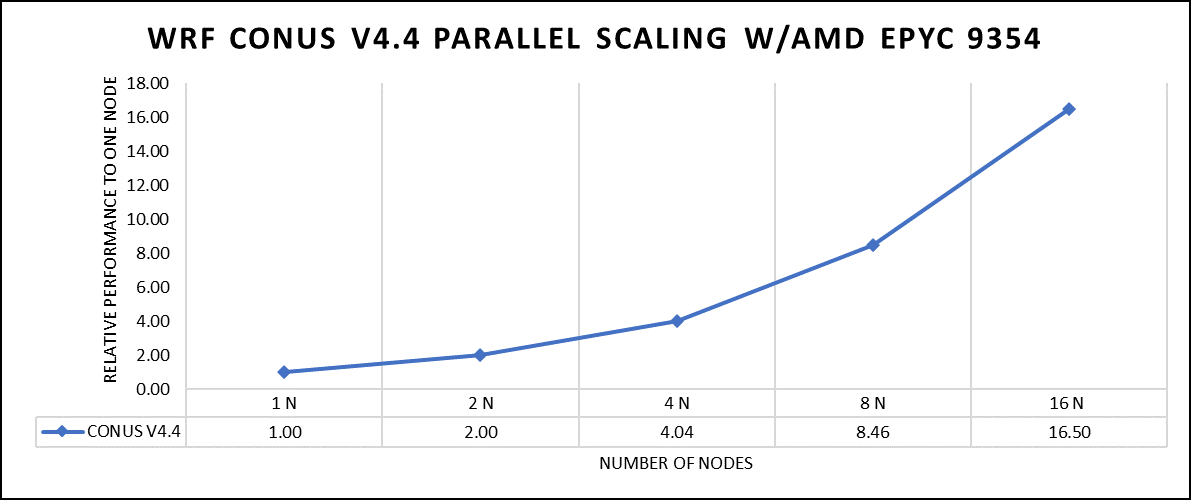
Figure 2: The scaling performance of the WRF CONUS dataset using the AMD EPYC 9354 processor, with a focus on performance compared to a single node.
The AOCC compiled binaries of WRF show linear scaling from a single node to sixteen nodes on 9354 processors for the new CONUS v4.4. For the best performance with WRF, the impact of the tile size, process, and threads per process should be carefully considered. Given that the application is constrained by memory and DRAM bandwidth, we have opted for the latest DDR5 4800 MT/s DRAM for our test evaluations. It is also crucial to consider the BIOS settings, particularly SubNUMA configurations, as these settings can significantly influence the performance of memory-bound applications, potentially leading to improvements ranging from one to five percent. For more detailed BIOS tuning recommendations, please see our previous blog post on optimizing BIOS settings for optimal performance.
Vertical – Molecular Dynamics | Application - LAMMPS
Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) is a powerful tool for HPC. It is specifically designed to harness the immense computational capabilities of HPC clusters and supercomputers. LAMMPS allows researchers and scientists to conduct large-scale molecular dynamics simulations with remarkable efficiency and scalability. In this study, we have worked on LAMMPS, 15 June 2023 version, which have been compiled with AOCC 4.0.0 with OPENMPI 4.1.4. For successful compilation and optimization with the AMD EPYC compilers, additional flags such as ' -O3 -znver4' have been added.
We opted for the non-default package, which offers optimized atom pair styles. We have also tried running some benchmarks which are not supported with default package to check the performance and scaling. Our performance metric for this benchmark is nanoseconds per day, where higher nanoseconds per day is considered a better result .
There are two factors that were considered when compiling data for comparison, the number of nodes and the core count. Figure 3 shows results of performance improvement observed on processor 9354 with 64 cores.
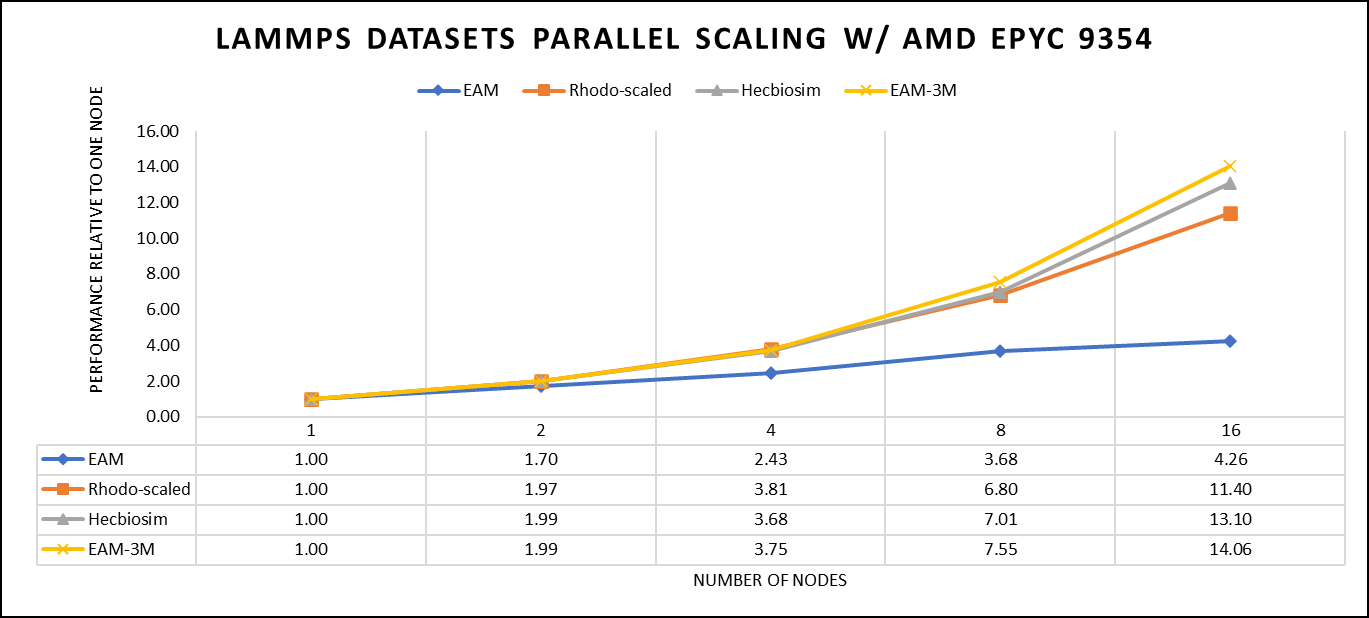
Figure 3: The scaling performance of the LAMMPS datasets using the AMD EPYC 9354 processor, with a focus on performance compared to a single node.
Figure 3 shows the scaling of different LAMMPS datasets. We see a significant improvement in scaling as we increased the atom size and step size. We have tested two datasets EAM and Hecbiosim with more than 3 million atoms and observed a better scalability as compared to other datasets.
Vertical – Molecular Dynamics | Application - GROMACS
GROMACS, a high-performance molecular dynamics software, is a vital tool for HPC environments. Tailored for HPC clusters and supercomputers, GROMACS specializes in simulating the intricate movements and interactions of atoms and molecules. Researchers in diverse fields, including biochemistry and biophysics, rely on its efficiency and scalability to explore complex molecular processes. GROMACS is used for its ability to harness the immense computational power of HPC, allowing scientists to conduct intricate simulations that unveil critical insights into atomic-level behaviors, from biomolecules to chemical reactions and materials. In this study, we have worked on GROMACS 2023.1 version, which have been compiled with AOCC 4.0.0 with OPENMPI 4.1.4. For successful compilation and optimization with the AMD EPYC compilers, additional flags such as ' -O3 -znver4' have been added.
We've curated a range of datasets for our benchmarking assessments. First, we included "water GMX_50 1536" and "water GMX_50 3072," which represent simulations involving water molecules. These simulations are pivotal for gaining insights into solvation, diffusion, and water's behaviour in diverse conditions. Next, we incorporated "HECBIOSIM 14K" and "HECBIOSIM 30K" datasets, which were specifically chosen for their ability to investigate intricate systems and larger biomolecular structures. Lastly, we included the "PRACE Lignocellulose" dataset, which aligns with our benchmarking objectives, particularly in the context of lignocellulose research. These datasets collectively offer a diverse array of scenarios for our benchmarking assessments.
Our performance assessment was based on the measurement of nanoseconds per day (ns/day) for each dataset, providing valuable insights into the computational efficiency. Additionally, we paid careful attention to optimizing the mdrun tuning parameters (i.e, ntomp, dlb tunepme nsteps etc )in every test run to ensure accurate and reliable results. We examined the scalability by conducting tests spanning from a single node to a total of sixteen nodes.
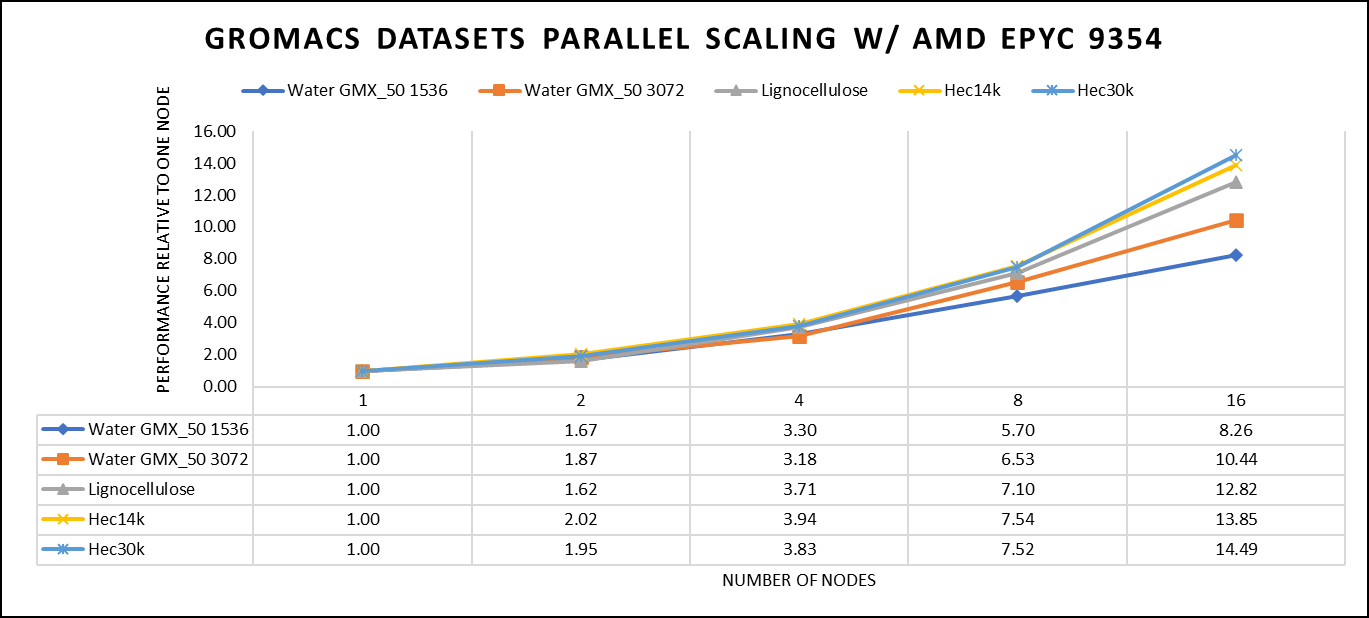
Figure 4: The scaling performance of the GROMACS datasets using the AMD EPYC 9354 processor, with a focus on performance compared to a single node.
For ease of comparison across the various datasets, the relative performance has been included into a single graph. However, it is worth noting that each dataset behaves individually when performance is considered, as each uses different molecular topology input files (tpr), and configuration files.
We were able to achieve the expected performance scalability for GROMACS of up to eight nodes for larger datasets. All cores in each server were used while running these benchmarks. The performance increases are close to linear across all the dataset types, however there is drop in larger number of nodes due to the smaller dataset size and the simulation iterations.
Vertical – Molecular Dynamics | Application – CP2K
CP2K is a versatile computational software package that covers a wide range of quantum chemistry and solid-state physics simulations, including molecular dynamics. It's not strictly limited to molecular dynamics but is instead a comprehensive tool for various computational chemistry and materials science tasks. While CP2K is widely used for molecular dynamics simulations, it can also perform tasks like electronic structure calculations, ab initio molecular dynamics, hybrid quantum mechanics/molecular mechanics (QM/MM) simulations, and more. In this study, we have worked on CP2K 2023.1 version, which have been compiled with AOCC 4.0.0 with OPENMPI 4.1.4. For successful compilation and optimization with the AMD EPYC compilers, additional flags such as ' -O3 -znver4' have been added.
In our study focusing on high-performance computing (HPC), we utilized specific datasets optimized for computational efficiency. The first dataset, "H2O-DFT-LS-NREP-4,6," was configured for HPC simulations and calculations, emphasizing the modeling of water (H2O) using Density Functional Theory (DFT). The appended "NREP-4,6" parameter settings were fine-tuned to ensure efficient HPC performance. The second dataset, "H2O-64-RI-MP2," was exclusively crafted for HPC applications and revolved around the examination of a system comprising 64 water molecules (H2O). By employing the Resolution of Identity (RI) method in conjunction with the Møller–Plesset perturbation theory of second order (MP2), this dataset demonstrated the significant computational capabilities of HPC for conducting advanced electronic structure calculations within a high-molecule-count environment. We examined the scalability by conducting tests spanning from a single node to a total of sixteen nodes.
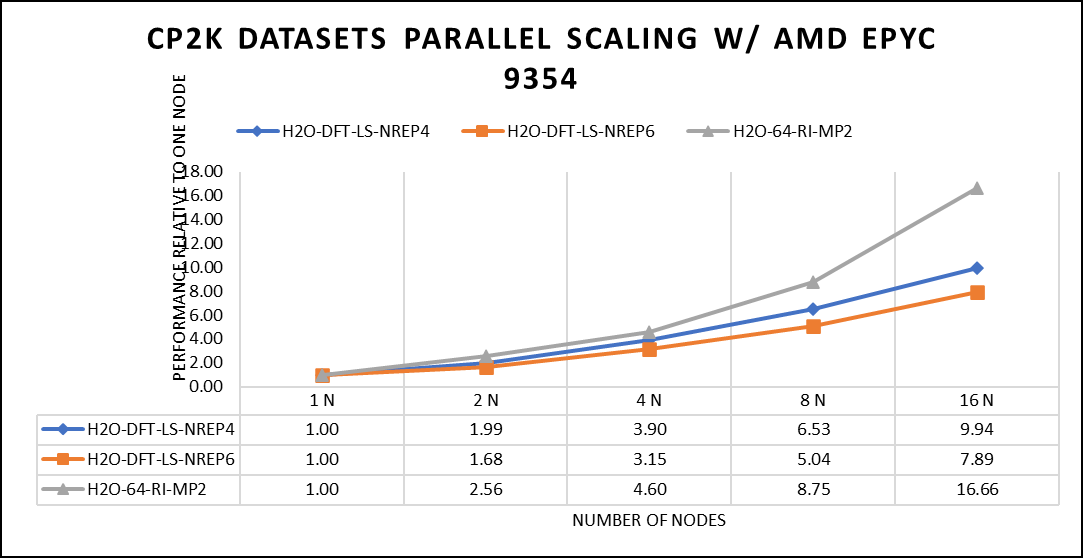
Figure 5: The scaling performance of the CP2K datasets using the AMD EPYC 9354 processor, with a focus on performance compared to a single node.
The datasets represent a single-point energy calculation employing linear-scaling Density Functional Theory (DFT). The system comprises 6144 atoms confined within a 39 Å^3 simulation box, which translates to a total of 2048 water molecules. To adjust the computational workload, you can modify the NREP parameter within the input file.
Our benchmarking efforts encompass configurations involving up to 16 computational nodes .Optimal performance is achieved when using NREP4 and NREP6 in Hybrid mode, which combines MPI (Message Passing Interface) and OpenMP (Open Multi-Processing). This configuration exhibits the best scaling performance, particularly on 4 to 8 nodes. However, it's worth noting that scaling beyond 8 nodes does not exhibit a strictly linear performance improvement. Above figure 5 depict outcomes when using Pure MPI, utilizing 64 cores with a single thread per core.
Conclusion
When considering CPUs with equivalent core counts, the earlier AMD EPYC processors can deliver performance levels like their Genoa counterparts. However, achieving this performance parity may require doubling the number of nodes. To further enhance performance using AMD EPYC processors, we suggest optimizing the BIOS settings as outlined in our previous blog post and specifically disabling Hyper-threading for the benchmarks discussed in this article. various workloads, we recommend conducting comprehensive testing and, if beneficial, enabling Hyper-threading. Additionally, for this performance study, we highly endorse the utilization of the Mellanox NDR 200 interconnect for optimal results.