




BWA-GATK Pipeline Performance on Dell EMC PowerEdge R6525 Server
Tue, 30 Mar 2021 18:34:08 -0000
|Read Time: 0 minutes
Overview
We’ve been speculating that AMD Milan with Zen3 cores which allows more cores to share the same L3 cache could perform better for Next Generation Sequencing (NGS) applications. Comparing to the predecessor AMD EPYC Rome, the number of cores sharing the L3 cache is doubled-up from 4 to 8 for the 64 core processor model. In addition to that, the cache (both L1 and L2) is upgraded with new prefetchers, and memory bandwidth is improved.
Since Milan and Rome share the same SP3 socket, Dell EMC PowerEdge R6525 was selected for the case study and able to minimize system-to-system variations. The test server configuration is summarized in Table 1.
Table 1. Tested compute node configuration
Dell EMC PowerEdge R6525 | |
CPU | Tested AMD Milan: 2x 7763 (Milan), 64 Cores, 2.45GHz – 3.5GHz Base-Boost, TDP 280W, 256 MB L3 Cache 2x 7713 (Milan), 64 Cores, 2.0GHz – 3.7GHz Base-Boost, TDP 225W, 256 MB L3 Cache 7543 (Milan), 32 Cores, 2.8GHz – 3.7 GHz Base-Boost, TDP 225W, 256 MB L3 Cache Tested AMD Rome: 7702 (Rome), 64 Cores, 2.0 GHz – 3.35 GHz Base-Boost, TDP 200W, 256 MB L3 Cache |
RAM | DDR4 256G (16Gb x 16) 3200 MT/s |
OS | RHEL 8.3 (4.18.0-240.el8.x86_64) |
Filesystem Network | Mellanox InfiniBand HDR100 |
Filesystem | Dell EMC Ready Solutions for HPC BeeGFS High Capacity Storage |
BIOS System Profile | Performance Optimized |
Logical Processor | Disabled |
Virtualization Technology | Disabled |
BWA | 0.7.15-r1140 |
Sambamba | 0.7.0 |
Samtools | 1.6 |
GATK | 3.6-0-g89b7209 |
The test data was chosen from one of Illumina’s Platinum Genomes. ERR194161 was processed with Illumina HiSeq 2000 submitted by Illumina and can be obtained from EMBL-EBI. The DNA identifier for this individual is NA12878. The description of the data from the linked website shows that this sample has a >30x depth of coverage, and it reaches ~53x.
Performance Evaluation
Characterizing steps in BWA-GATK Pipeline
In a typical BWA-GATK pipeline, there are multiple steps, and each step consists of various applications which behave distinctively. As shown in Table 3, the applications in some steps do not support multi-threading operations. These steps are problematic since there are only a few ways to improve performance.
Table 2. The steps in BWA-GATK pipeline and tools
Steps | Applications | Multi-threading support |
Mapping & Sorting | BWA, samtools, sambamba | Yes |
Mark Duplicates | Sambamba | Yes |
Generate Realigning Targets | GATK RealignerTargetCreator | Yes |
Insertion/Deletion Realigning | GATK IndelRealigner | No |
Base Recalibration | GATK BaseRecalibrator | Yes |
Haplotypercaller | GATK HaplotypeCaller | Yes |
Genotype GVCFs | GATK GenotypeGVCFs | Yes |
Variant Recalibration | GATK VariantRecalibrator | No |
Apply Variant Recalibration | GATK ApplyRecalibration | No |
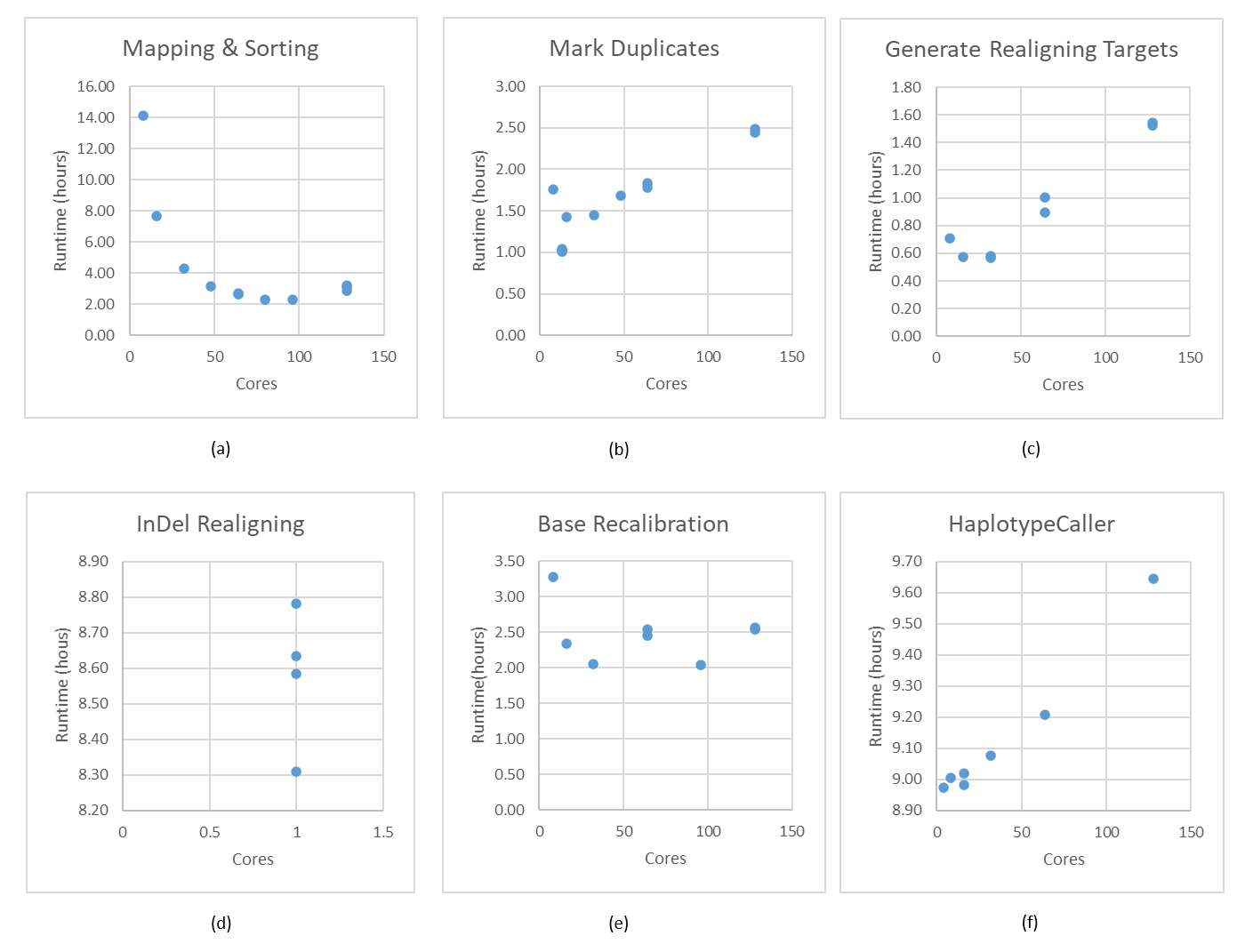
Single thread applications, especially Variant Recalibration and Apply Variant Recalibratrion steps show no runtime variation due to the deterministic algorithm and the inputs for these steps are small. Hence, these two steps are not reported in Figure 1. The first step, Mapping & Sorting scales as the number of cores increases (Figure 1, (a)). Also, Genotype GVCFs is not included in Figure 1 although it supports multi-threading operation for a similar reason.
Burrows-Wheeler Aligner (BWA) is one of the most popular short sequence aligners for non-gapped aligning analysis. BWA scales well until 32 cores, and CPU usage drops down dramatically after 32 cores. The runtime improvement becomes marginal with higher core numbers greater than 32. Using more than 80 cores for this step is the wasting of resources.
Sambamba which is compatible with Picard is used for marking duplicate reads. The behavior of sambamba is plotted in Figure 1, (b). Due to the highly parallelized nature of design, the memory consumption increases to create more hash tables for additional threads. Amazingly, 50x human whole genome sequence (WGS) is not big enough to use more than 13 cores for the well-designed software.
After Mark Duplicates step, Genome Analysis Tool Kit, hence GATK, written in Java plays a critical role in performance measurement and creating answers. These steps do not scale at all as shown in Figure 1, (c) (d) (e) and (f). A better approach will be discussed in future work to handle the misbehavior in multi-core and multi-socket environments.
Single Sample Performance
Socket to Socket Comparison
This test is not a fair comparison since the majority of steps will not take advantage of using all the cores except 7543 with 32 cores. However, this comparison will help to decide which CPU could be best for the throughput test.
Table 3 summarizes the overall runtimes for BWA-GATK pipeline, and it is hard to say which one is better in terms of total runtimes. A lot more tests are required to differentiate the performance differences in GATK steps. Also, the results from 7502 and 7402 were from the previous tests with different environments.
The mapping & Sorting step is the only step that we can peak the true performance variations across different CPUs in Table 3. A rough estimation of performance improvement from 7702 to 7763 is 7% while the performance gain is 5% from 7702 to 7713.
Surprisingly, the Base Recalibration step showed similar results as the Mapping & Sorting step, which is 8% and 3% improvement.
Table 3. BWA-GATK performance comparisons between Milan and Rome. The number of cores used for the test is parenthesized.
Steps | Runtime (hours) | |||||
AMD 7763 64c 2.45GHz | AMD 7713 64c 2.0GHz | AMD 7543 32c 2.8GHz | AMD 7702 64c 2.0GHz | AMD 7502 32c 2.5GHz | AMD 7402 24c 3.0GHz | |
Mapping & Sorting | 2.44 (64) | 2.49 (64) | 3.69 (32) | 2.63 (64) | 4.68 (32) | 5.73 (24) |
Mark Duplicates | 1.07 (13) | 1.10 (13) | 1.01 (13) | 1.01 (13) | 0.93 (13) | 0.94 (13) |
Generate Realigning Targets | 0.55 (32) | 0.56 (32) | 0.50 (32) | 0.58 (32) | 0.45 (32) | 0.44 (32) |
Insertion/Deletion Realigning | 8.73 (1) | 9.13 (1) | 7.73 (1) | 8.78 (1) | 8.30 (1) | 8.21 (1) |
Base Recalibration | 2.27 (32) | 2.38 (32) | 2.17 (32) | 2.46 (32) | 2.52 (32) | 2.67 (24) |
Haplotypercaller | 10.20 (16) | 10.57 (16) | 9.15 (16) | 9.02 (16) | 9.33 (16) | 9.05 (16) |
Genotype GVCFs | 0.02 (32) | 0.02 (32) | 0.01 (32) | 0.02 (32) | 0.01 (32) | 0.01 (24) |
Variant Recalibration | 0.31 (1) | 0.20 (1) | 0.17 (1) | 0.12 (1) | 0.21 (1) | 0.13 (1) |
Apply Variant Recalibration | 0.01 (1) | 0.01 (1) | 0.01 (1) | 0.01 (1) | 0.01 (1) | 0.01 (1) |
Total Runtime (hours) | 25.59 | 26.47 | 24.44 | 24.64 | 26.46 | 27.25 |
Multiple Sample Performance - Throughput
A typical way of running an NGS pipeline is to process multiple samples on a compute node and use multiple compute nodes to maximize the throughput. However, this time the tests were performed on a single compute node due to the limited number of servers available at this moment.
The current pipeline invokes a large number of pipe operations in the first step to minimize the amount of writing intermediate files. Although this saves a day of runtime and lowers the storage usage significantly, the cost of invoking pipes is quite heavy. Hence, this limits the number of concurrent sample processing. Typically, a process silently fails when there is not enough resource left to start an additional process.
However, the failures experiencing during this study are quite different from the previous observations. 10 pipelines started in R6525 with 2x 7763 sustain only 6 pipelines on average with the 50x human WGS. Four pipelines are failed with the broken pipes error which suggests some sort of file operation. Current BeeGFS storage for the test is designed for high capacity, theoretical sequential write bandwidth of 25 GB/s. However, roughly 16 GB/s is achievable where there is not heavy usage loaded on this storage in a shared storage environment. This is not an ideal environment for any benchmark practice; however, the results here are quite helpful to see what the performance of these systems looks like in a real life.
As shown in Table 4, the maximum number of samples that can be processed at the same time is around 4 or 5, and the ~ 4.79 50x human whole genomes per day throughput is achievable with the current environment.
Table 4. Throughput test for Milan 7763
Steps | Runtime (hours) | ||||
1 Sample | 2 Samples | 4 Samples | 6 Samples | 10 Samples | |
Number of Samples Failed | 0 | 0 | 0 | 1 | 4 |
Mapping & Sorting | 2.44 | 2.91 | 4.33 | 5.86 | 8.33 |
Mark Duplicates | 1.07 | 1.40 | 1.69 | 1.31 | 5.51 |
Generate Realigning Targets | 0.55 | 0.88 | 1.77 | 0.50 | 2.07 |
Insertion/Deletion Realigning | 8.73 | 8.97 | 8.92 | 8.92 | 9.70 |
Base Recalibration | 2.27 | 2.50 | 2.79 | 3.26 | 3.67 |
Haplotypercaller | 10.20 | 10.57 | 10.27 | 9.91 | 9.96 |
Genotype GVCFs | 0.02 | 0.11 | 0.10 | 0.10 | 0.15 |
Variant Recalibration | 0.31 | 0.25 | 0.20 | 0.21 | 0.36 |
Apply Variant Recalibration | 0.01 | 0.02 | 0.01 | 0.01 | 0.03 |
Total Runtime (hours) | 25.59 | 27.62 | 30.08 | 30.08 | 39.79 |
Genomes per day | 0.94 | 1.74 | 4.79 | 3.99 | 3.62 |
Conclusion
The field of NGS data analysis has been moving fast in terms of data growth and data variations. However, the community has not been done much work adapting new technologies available such as accelerators. Instead of improving the quality of codes, the community is faced with analyzing the data without multi-thread processing since GATK version 4 and up does not support multi-threading anymore while the number of cores in a CPU increases fast.
It is time that users need to think about how this problem can be tackled. One simple way to avoid this problem is to perform data-level parallelization. Although the decision-making of when to split data is pretty hard, it is certainly tractable with careful interventions in an existing BWA-GATK pipeline without diluting statistical power with a sheer number of data. If each smaller data chunk goes through an individual pipeline on each core and is merged at the end, it could be possible to achieve better performance on a single sample. This performance gain could lead to higher throughput if the overall runtime reduces significantly.
Nonetheless, Milan 7763 or 7713 are an excellent candidate to cover both current multi-threading-based pipelines and future data-level parallelization driven pipelines with more available cores.