




Molecular Dynamics Simulations with Dell EMC PowerEdge XE8545 Server and NVIDIA A100
Wed, 02 Jun 2021 19:37:48 -0000
|Read Time: 0 minutes
Overview
Over the past decade, graphics processing units, or GPUs, have become popular in scientific computing because of their great ability to exploit a high degree of parallelism. NVIDIA has a handful of life sciences applications optimized and run on their general-purpose GPUs. Unfortunately, these GPUs can only be programmed with CUDA, OpenACC, and the OpenCL framework. Most members of the life sciences community are not familiar with these frameworks, and so few biologists or bioinformaticians can make efficient use of GPU architectures. However, GPUs have been making inroads into the molecular dynamics simulation (MDS) field since MD was developed in the 1950s. MDS requires heavy computational work to simulate biomolecular structures or their interactions.
In this blog, we tested two MDS applications; NAMD, and LAMMPS using the Dell EMC PowerEdge XE8545 server with NVIDIA A100 GPUs. Since the XE8545 server does not support NVIDIA V100 GPU, we can roughly estimate the performance boost with the A100 from our previous tests.
These two applications are free and open-source parallel MD packages designed for analyzing the physical movements of atoms and molecules.
The test server configuration is summarized in the following table.
Dell EMC PowerEdge XE8545 | |
CPU | 2x 7713 (Milan), 64 Cores, 2.0 GHz – 3.7 GHz Base-Boost, TDP 225 W, 256 MB L3 Cache |
RAM | DDR4 1024 GB (32 x 32 GB) 3200 MT/s |
Operating system | RHEL 8.3 (4.18.0-240.el8.x86_64) |
Filesystem network | Mellanox InfiniBand HDR100 |
Filesystem | Dell EMC Ready Solutions for HPC BeeGFS High Capacity Storage |
BIOS system profile | Performance Optimized |
Logical processor | Disabled |
Virtualization technology | Disabled |
Accelerator | 4 x A100-40 GB SXM4 |
Cuda/Toolkit | 11.2 |
OpenMPI | 4.1.1 |
NAMD | NAMD_Git-2021-04-01_Source |
LAMMPS | Stable version (29 Oct 2020) |
Performance Evaluation
NAMD
Nanoscale Molecular Dynamics (NAMD) is open-source software for molecular dynamics simulation written in a CHARMM parallel programming model and is designed for high-performance simulation of large biomolecular systems.
NAMD was built with the NAMD_Git-2021-04-01_Source source code on GCC 11.1 and CUDA 11.2. For our tests, we used two sets of data; 1.06 million-atoms of the Satellite Tobacco Mosaic Virus (STMV) system, and the HECBioSim3000k-atom system, which is a pair of 1IVO and 1NQL hEGFR tetramers.
Figure 1 shows the performance of 4x A100 GPUs with the STMV dataset. NAMD uses ++p options to specify the number of worker threads, and as recommended, is equal to the total number of cores minus the total number of GPUs. However, the number of total cores in the Milan Eypc 7003 family of processors, such as the Eypc 7713 that is used in the testing system, does not follow the generic recommendation. It seems to be around 79 to 90 cores. The optimal number of cores depends on the data size. Close to 9-nanosecond simulations (ns) per day performance is a significant performance gain from the NVIDIA V100 tests that we ran previously. It is difficult to say the performance gain is the sole contribution of the new A100 GPUs because the comparison of the 16 GB V100 on the Intel Skylake platform to the 40 GB A100 on the AMD Milan platform may not be valid.
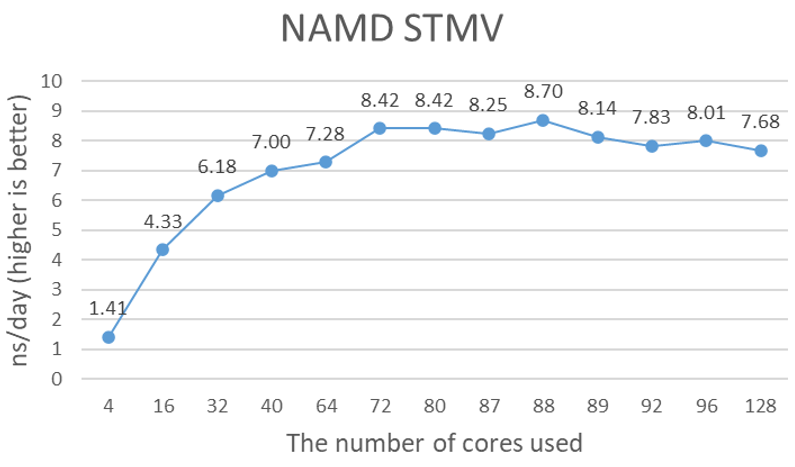
Figure 1. Estimated simulation time per day with 4x NVIDIA A100 GPUs
The purpose of an additional test with 3 million atom protein tetramers is to confirm that the STMV test results are not artificial due to the relatively small icosahedron structure of SMTV, and the partial simulation of assembly and disassembly processes. Figure 2 shows the nanosecond simulations per day plot for 3000k-atom data. 2.1 ns/day seems to be close to the maximum performance with 64 cores.
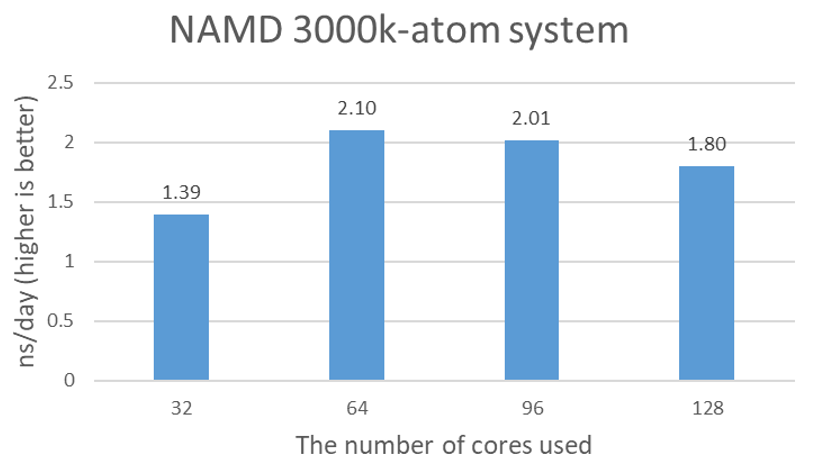
Figure 2. Estimated simulation time per day with 4x NVIDIA A100 GPUs
LAMMPS
Large-scale Atomic/Molecular Massively Parallel Simulator, or LAMMPS, is a classical molecular dynamics code and has potentials for solid-state materials (metals and semiconductors), soft matter (biomolecules and polymers), and coarse-grained or mesoscopic systems. LAMMPS can model atoms, or can be used as a parallel particle simulator at the atomic, meso, or continuum scale. LAMMPS runs on single processors, or in parallel using message-passing techniques and spatial decomposition of the simulation domain. LAMMPS was built with GCC 11.1, OpenMPI 4.1.1, and CUDA 11.2 from the source. The 465k-atom system was selected from HECBioSim.
As shown in Figure 3, LAMMPS scales well over the number of A100s. With 4x A100 GPUs, a 8.4 ns/day simulation is achievable.
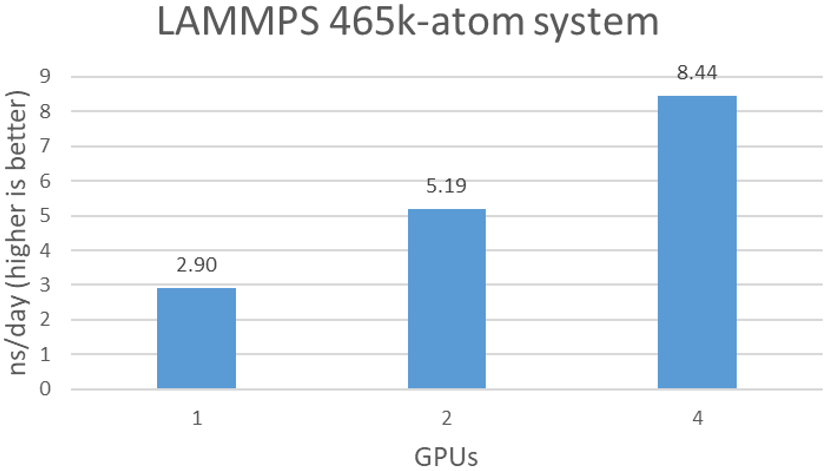
Figure 3. Estimated simulation time per day with various number of BPUs
Conclusion
Although it is not possible to compare the performance of the A100 and the V100 from this study, the Milan CPUs and A100 show a strong synergy between more cores with better and faster GPUs. Running NAMD and LAMMPS on the XE8545 with the A100 can deliver a better performance than a system with the V100.