
BWA-GATK Germline variant calling pipeline
BWA-GATK Germline variant calling pipeline
-
While BWA has been stable for many years, some radical modifications are made to GATK version 4 and up. While the versions of GATK before version 4 support multithreading, versions 4 and up support only single-thread operation. To remedy this shortcoming, Spark version of GATK is also provided; however, it is mostly experimental.
For benchmarking GATK v3.6, Dell Technologies used the same pipeline for all the previous benchmarks. However, two different versions of pipelines for GATK v4.2 are created based on germline cohort data analysis pipeline as shown in Figure 5.
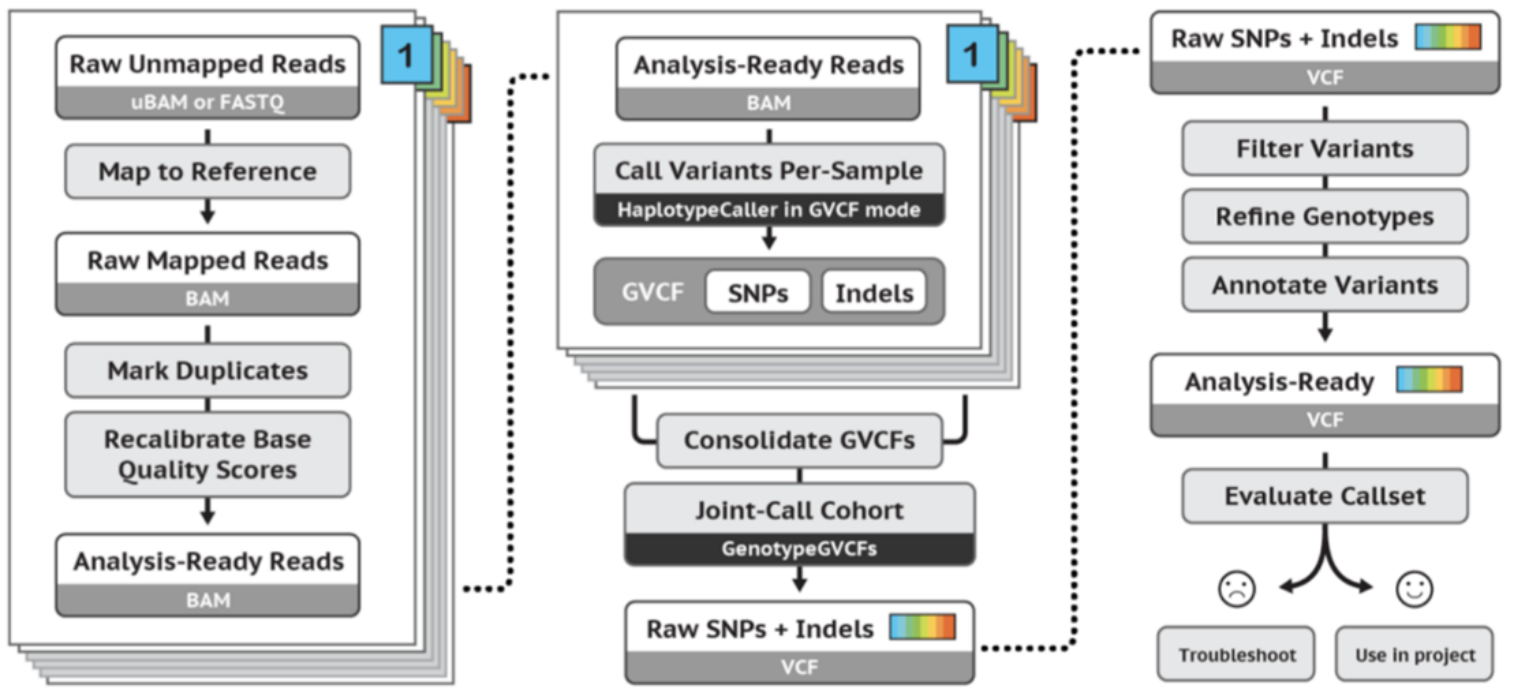
Figure 5. Main steps for Germline Cohort Data
BWA-GATK v3.6 pipelines
This pipeline is designed to perform germline variant calling for benchmark purposes. Steps in the pipeline and their applications are listed in Table 6.
Step 1 consists of three applications piped together. An output from BWA is directly passed to Samtools for the file conversion from SAM to BAM. And the converted output from Samtools feeds to Sambamba for sorting the sequence reads by their chromosome locations. This piping technique can save writing a large file twice; however, it can deplete the multitasking capability of the CPU easily if many samples are processed through this pipeline. There is an upper number of samples concurrently processed with piping. Otherwise, the total number of cores in a system is the maximum number of samples that can be run together.
Without a proper cohort dataset, it is not possible to obtain a reliable runtime from Step 7, Joint-Call Cohort. It is worth noting that the results from Step 7 are not realistic as Dell Technologies tested with a single sample. The runtime of Step 7 will grow according to the size of the cohort.
Table 6. Steps in the tested BWA-GATK v3.6 pipeline
Step
Operation
Applications
1
Align and Sort
BWA, Samtools, Sambamba
2
Mark and Remove Duplicates
Sambamba
3
Generate Realigning Targets
GATK - RealignerTargetCreator
4
Realign around Insertion and Deletion
GATK - IndelRealigner
5
Recalibrate Base
GATK - BaseRecalibrator
6
Call Variants
GATK - HaplotypeCaller
7
Consolidate GVCFs
GATK – GenotypeGVCFs
8
Recalibrate Variants
GATK – VariantRecalibrator
9
Apply Variant Recalibrations
GATK - ApplyRecalibration
BWA-GATK v4.2 pipeline
With GATK version 4 and up, the GATK steps are simplified dramatically. Especially, Step 4 is removed in Table 6 above saving more than seven hours of runtime. However, GATK version 4 and up do not support multiple threading, which increases the runtime nearly three times more than the BWA-GATK v3.6 pipeline.
Table 7. Steps in the tested BWA-GATK v4.2 pipeline
Step
Operation
Applications
1
Align and Sort
BWA, Samtools, Sambamba
2
Mark and Remove Duplicates
Sambamba
3
Recalibrate Base and Generate BQSR
GATK - BaseRecalibrator
4
Apply BQSR
GATK - ApplyBQSR
5
Call Variants
GATK - HaplotypeCaller
6
Consolidate GVCFs
GATK - GenotypeGVCFs
BWA-GATK v4.2 data parallelization pipeline
Due to the lack of multithreading support in GATK v4.2, the processing time of a single sample could increase three times more than the runtime from GATK v3.6. It is not fair to compare the runtimes between two different versions of GATK since they are processing the data in different ways. However, overall runtimes of a v4.2 pipeline dramatically increase, and it is obvious that it will cause a severe bottleneck in the data processing.
Adding data parallelization steps is not simple because it can cause data dilution at the statistical steps in Steps 3 through 6. Without data parallelization, these steps will see the entire data provided in Step 1. For example, an input sequence read data is 50x WGS, these steps will use at around 50 examples per chromosome location. However, if the input is split into 50 chunks for the data parallelization, these steps essentially convert the 50x data to 1x WGS data. This splitting will be the main cause of unreliable results. The idea of proposed data parallelization is adding the DP 2 step to merge aligned data. Then, splitting the data one more time to keep all reads from a similar region of chromosomes. This parallelization is possible because the sequence reads are sorted based on where these reads are matched on chromosomes. However, splitting data into multiple files requires a significant operational time since writing multiple small files is involved. At steps DP 3 and 4, count the number of reads successfully aligned in Step 1. Then generate index files to record what are the chromosome location starts and ends that equally divide the number of reads. Step DP 4 generates multiple interval files to pass on subsequent steps, Steps 3 through 6 allow steps to operate only in the region specified in an interval file. Once concurrent steps generate final outputs at Step 6, these results are stitched together at the DP 5 step.
Table 8. Steps in the tested BWA-GATK v4.2 data parallelization pipeline
Operation
Applications
DP 1
Split FASTQ file
Customized
1
Align and Sort
BWA, Samtools, Sambamba
2
Mark and Remove Duplicates
Sambamba
DP 2
Merge BAM files
Customized
DP 3
Count the number of aligned reads
Customized
DP 4
Generate Interval files
Customized
3
Recalibrate Base and Generate BQSR
GATK - BaseRecalibrator
4
Apply BQSR
GATK - ApplyBQSR
5
Call Variants
GATK - HaplotypeCaller
6
Consolidate GVCFs
GATK - GenotypeGVCFs
DP 5
Merge VCF files
Customized
Note: More data parallelization steps are labeled as DP.